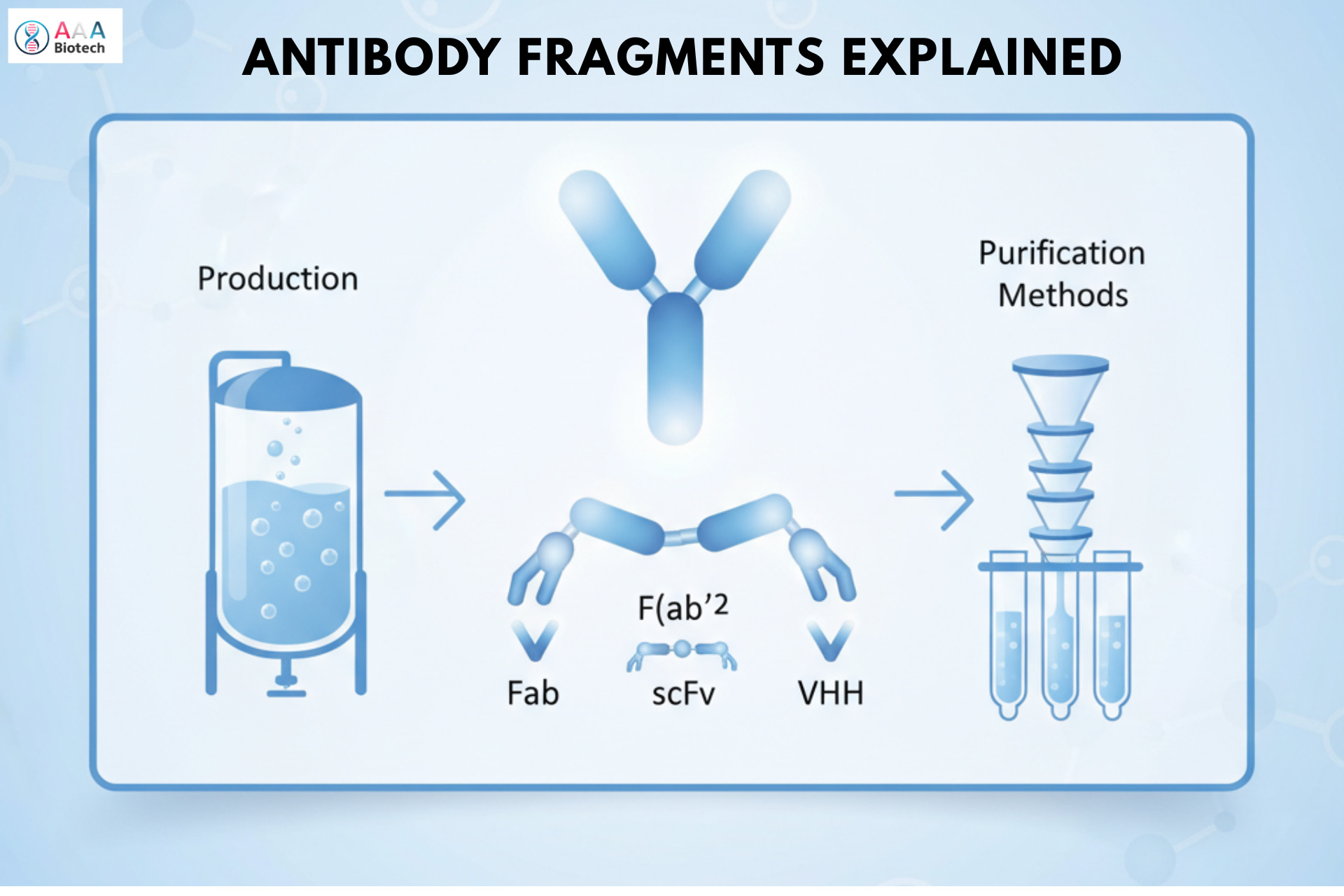
Understanding Antibody Glycosylation, Its Types, Importance, & Profound Impact on Biotechnological Research
The Definitive Western Blot Guide: Steps, Uses, and Its Essential Role in Research
In this Article
All of the products listed in AAA Biotech’s catalog are strictly for research-use only (RUO).

Key Takeaways
- The Western blot is the gold standard for identifying specific proteins using an antibody-antigen interaction.
- The technique involves three core steps: Separation (SDS-PAGE), Immobilization (Transfer to a membrane), and Identification (Immunoassay).
- Meticulous sample preparation (with fresh protease/phosphatase inhibitors) is critical to prevent protein degradation and artifacts.
- Protein quantification (e.g., BCA assay) ensures equal loading for accurate semi-quantitative analysis.
- PVDF membranes are durable for stripping and re-probing; nitrocellulose has lower autofluorescence.
- Blocking the membrane with non-fat milk or BSA prevents high background noise from non-specific antibody binding.
- The primary antibody dilution is the single most important factor for achieving a clean, specific signal.
- Fluorescent detection allows for multiplexing and more robust total protein normalization.
The Western blot, often referred to as immunoblotting, stands as a cornerstone
technique in molecular biology and proteomics. It is the gold standard for
separating and identifying specific proteins from a complex mixture, utilizing
the highly specific interaction between an antibody and its target antigen.
Understanding the precise Western blotting principle and mastering each
meticulous step of the Western blot procedure is fundamental to generating
robust, publishable scientific data.
This comprehensive guide details the entire Western blot protocol, from initial sample preparation to final analysis, offering essential technical rationales and optimization strategies necessary for reliable research results.
Defining the Immunoblotting Principle
The term “Western blot” refers to a powerful analytical technique used for
protein detection. It is a four-stage process that allows researchers to analyze
complex biological samples, such as cell lysates or tissue homogenates. The key
feature of this Western blot method is that it uses an antibody to specifically
detect its corresponding antigen (the protein of interest).
The power of Western blot lies in its ability to generate both qualitative data (confirming the presence or absence of a protein) and semi-quantitative data (providing an estimation of the relative abundance of a protein).
The foundational success of Western blot analysis relies on linking three distinct biophysical techniques in a precise sequence:
- Separation: Macromolecules are physically separated,
typically based on size, using gel electrophoresis.
- Immobilization: The separated proteins are fixed onto a
stable, solid support matrix (the membrane).
- Identification: A highly specific immunoassay uses antibodies to tag and visualize the target protein against the backdrop of thousands of other cellular components.
Since the reliability of the result depends on the integrity of every phase, meticulous execution of the Western blot procedure is crucial for accurate biological conclusions.
Phase I – Optimized Sample Preparation and Separation
Success in the Western blotting technique begins long before the gel is cast.
The integrity of the final data is directly dependent upon the quality of the
initial protein extract.
Critical Western Blot Sample Preparation
The primary objective of Western blot sample preparation is to efficiently lyse the source material (cells or tissue) and solubilize all proteins while preserving their chemical state (e.g., phosphorylation status) and preventing degradation.
01. Lysis and Preservation
Preparation must be executed quickly and stably at cold temperatures (on ice) to
minimize enzymatic activity.
The critical ingredient is the lysis buffer, which must be supplemented with inhibitors. The lysis process breaks down cellular compartments, releasing active enzymes such as proteases (which cleave proteins) and phosphatases (which remove phosphate groups). If these enzymes are active, the protein of interest can be destroyed or modified, generating artifactual results (like smearing or non-specific bands). Therefore, the inclusion of fresh protease and phosphatase inhibitors (such as Halt Cocktail or specialized tablets) is non-negotiable, particularly when investigating signaling pathways or measuring protein modifications. For example, EDTA can also be added to the buffer to inhibit metalloproteases.
02. Extraction Techniques
The appropriate lysis technique depends heavily on the source material:
- Cultured Cells (e.g., HEK293): Often involves placing the
plate on ice, washing with cold PBS, adding lysis buffer, and gently scraping or
agitating the cells.
- Tissues or Hardy Cells (e.g., Bacteria, Yeast): Requires harsher, mechanical methods to fully disrupt the robust structure. These can include sonication (using ultrasonic waves), French press (high-pressure shear forces), or grinding the tissue to a fine powder using a mortar and pestle cooled with liquid nitrogen.
03. Sample Loading and Quantification
Prior to loading, protein concentration must be determined (typically using BCA
or Bradford assays) to ensure equal loading across all lanes, which is essential
for accurate semi-quantitative Western blot analysis.
Total protein loading per lane typically ranges from 10 to 50 μg. However, loading too much protein (high protein concentration) is a common cause of high background, non-specific binding, and streaky blots.
For many applications, 30 μg of protein per lane is the recommended starting point to yield streak-free and well-separated bands. For low-abundance proteins, researchers may need to increase loading or pre-concentrate the target using techniques such as immunoprecipitation (IP) or affinity column chromatography (e.g., using HIS tags).
04. Denaturation and Reduction
Most Western blot protocols involve separating proteins in a reduced and
denatured state to separate them solely based on mass. This involves preparing
the protein sample with:
- SDS/LDS Sample Buffer: To denature the protein and coat it
with a uniform negative charge.
- Reducing Agent: Such as DTT (Dithiothreitol) or
beta-Mercaptoethanol to break internal disulfide bonds.
- Heat: Samples are typically heated for 10 minutes, although
boiling for 5–10 minutes may be used for integral membrane proteins.
- Tracking Dye: Bromophenol blue is added to visualize protein migration, as it moves faster than the proteins.
Protein Separation: SDS-PAGE
The first major Western blot phase is separation via gel electrophoresis. Polyacrylamide gel electrophoresis (PAGE) is used to characterize individual proteins or examine complex samples. The most common form is Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE), which separates proteins primarily according to their mass after the proteins are bound by the ionic detergent SDS.
To achieve optimal resolution, several factors must be considered:
- Gel Uniformity: Whether using precast or self-cast gels,
uniformity is essential to prevent distortion in protein migration.
- Running Conditions: Running the gel at excessive voltage
must be avoided to prevent thermal distortion, known as the "dye-front smiling"
effect.
- Buffer System: Different buffering systems are available, such as Tris-glycine, Bis-Tris, or Tris-acetate, and the selection depends on the protein size and the desired separation profile.
Phase II – Transfer and Membrane Strategy
Following separation, the proteins must be transferred from the fragile gel
matrix to a durable, solid support for subsequent immunological probing. This is
the "blotting" component of the western blotting technique.
Electrophoretic Transfer (The Blotting Step)
The most efficient Western blot method for transfer is electroelution, or electrophoretic transfer, which utilizes the proteins' electrophoretic mobility.
The gel is placed in direct contact with a piece of membrane (nitrocellulose or PVDF) in a "sandwich" assembly, submerged between electrodes in a conducting solution. When an electric field is applied, the proteins move out of the gel and become tightly attached to the membrane surface.
Transfer Optimization and Verification
Transfer parameters (time and voltage) are not universal and must be optimized
based on the target protein's molecular weight (MW).
- Small Proteins: Proteins under 20 kDa move quickly and can
run completely through the membrane. Adjustments, such as reducing transfer time
or using a smaller pore size membrane, may be necessary.
- Large Proteins: Proteins over 100 kDa may require longer transfer times or specialized buffer additives to efficiently elute from the gel.
After the transfer, efficiency must be checked. A common reversible staining
reagent is Ponceau S stain, which quickly assesses the total protein transferred
onto the membrane. This check is critical not only to confirm that the transfer
occurred but to visualize if proteins were transferred uniformly across the full
range of molecular weights.
Common transfer mistakes include a poorly fitted or loose sandwich, and running the transfer too quickly thereby generating heat (which can compromise the entire process). Running the transfer at a lower voltage for a longer time is often recommended to maintain efficiency and integrity.
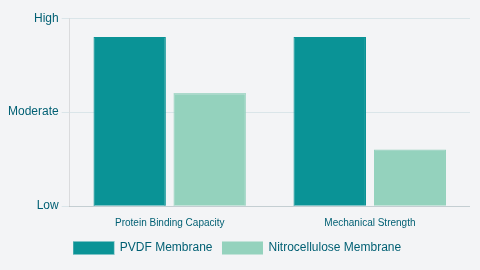
Choosing Your Matrix: Nitrocellulose vs. PVDF Membrane
The choice of membrane material is also a critical decision in the Western blot procedure, impacting robustness, sensitivity, and detection compatibility. The two main matrices are nitrocellulose and polyvinylidene difluoride (PVDF).
The key differences relate to binding capacity, mechanical strength, and performance in advanced detection systems.
| Feature | PVDF Membrane | Nitrocellulose Membrane | Implications for Research |
|---|---|---|---|
| Protein Binding Capacity | High | Moderate | Higher sensitivity, preferred for low-abundance proteins |
| Mechanical Strength | High Durability | Low (Prone to tearing) | Essential for stripping and reprobing multiple times |
| Hydrophobicity | Highly Hydrophobic | Easily Hydrated | Requires methanol activation/wetting step. Preferred for hydrophobic membrane proteins. |
| Standard Autofluorescence | High | Low | Standard type is unsuitable for fluorescence detection, requiring specialized Low Fluorescence PVDF |
Table 01: Membrane Selection Guide for the Western Blotting Technique
PVDF membranes offer superior mechanical strength and chemical resistance, making them ideal when the researcher plans to strip the antibodies and re-probe the blot multiple times. Their higher protein-binding capacity also provides increased sensitivity, which is beneficial for detecting low-abundance targets.
Nitrocellulose membranes are easily hydrated and feature instantaneous protein binding.
Standard PVDF membranes exhibit high autofluorescence, so if the final Western blot detection methods rely on fluorescence (for multiplexing), then specialized low-fluorescence PVDF must be used, or the researcher must opt for nitrocellulose, accepting the trade-off in mechanical durability. Finally, membrane pore size must also be selected, as smaller pore sizes (e.g., 0.2 μm ) are needed to effectively capture very small proteins and prevent them from passing completely through the membrane.
Phase III – Immunological Detection and Reagents
This phase is the core immunoassay component of the Western blotting technique,
where specific immunological reagents identify the target protein.
Blocking Nonspecific Sites
After transfer, the membrane contains proteins only in the areas where the separated bands are located, leaving vast empty areas of the synthetic membrane surface. These unoccupied regions are highly attractive to proteins, including the expensive primary and secondary antibodies used later in the western blot procedure.
Blocking is the process of saturating these empty sites with inert protein (the blocking agent) to prevent any nonspecific binding of the antibodies, thereby reducing background noise and significantly improving the signal-to-noise ratio.
Selecting the Optimal Blocking Reagent
The choice of blocking buffer is highly specific to the experiment and often
dictates the success of the Western blot analysis. The two most common western
blot reagents for blocking are non-fat milk and Bovine Serum Albumin (BSA).
Antibody Incubation (Primary and Secondary)
The Western blot method typically employs the indirect detection scheme, involving two antibody incubation steps separated by rigorous washing steps.
Primary Antibody Incubation
The blocked membrane is incubated with an unlabeled primary antibody that is
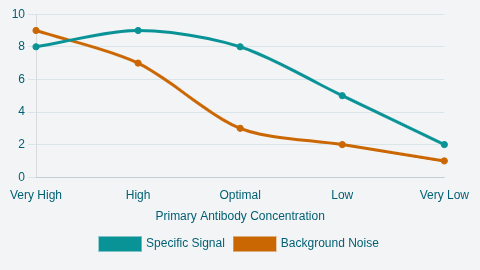
specific to the protein of interest. The concentration of the primary antibody
is the single most important factor for achieving a clean signal. Antibodies are
used in highly dilute solutions, often ranging from 1/100 to 1/500,000.
Optimization of this dilution is critical. If the manufacturer recommends a starting point (e.g., 1:1000), researchers should test a range that brackets this dilution, such as 1:250, 1:500, 1:1000, 1:2000, and 1:4000. Using too high a concentration results in excessive nonspecific binding, creating a dark, high-background blot.
Secondary Antibody Incubation
Following washing, the membrane is incubated with an enzyme- or
fluorophore-conjugated secondary antibody. This antibody is designed to bind to
the constant region of the primary antibody, based on the host species in which
the primary antibody was raised (e.g., Anti-Rabbit IgG detects a primary
antibody raised in a rabbit).
The secondary antibody is conjugated with a detectable label. Common labels include enzymes such as Horseradish Peroxidase (HRP) or Alkaline Phosphatase (AP), or fluorescent probes. These labels provide the signal amplification necessary for visualization.
Washing Steps
Washing is a vital Western blot step that actively removes unbound and non-specifically bound antibodies and other reagents. If washing is insufficient, a high background will result, leading to a dark or speckled blot that obscures the specific target signal.
The most common wash buffers are Tris-buffered saline (TBS) and phosphate-buffered saline (PBS). These are often supplemented with a mild detergent, typically Tween 20 (0.05% to 0.5%), to help dislodge non-specifically bound material.
It is important to note that TBS should be preferentially used over PBS when the detection system utilizes AP-conjugated secondary antibodies or when detecting phosphorylated proteins, as the phosphate ions in PBS can inhibit AP activity or interfere with phospho-detection chemistries. The washing steps must be meticulous and timed precisely to maximize the signal-to-noise ratio.
Phase IV – Detection, Analysis, and Troubleshooting
The final Western blot procedure culminates in the generation and capture of a
detectable signal that allows for analysis and quantification.
Western Blot Detection Methods
The choice of the detection methods dictates the sensitivity, dynamic range, and potential for multiplexing.
01. Enzymatic Detection (Chemiluminescence)
This is the most widely used and sensitive system. Enzymes, typically HRP or AP,
react with a specific substrate to produce light (chemiluminescence). The light
output is captured using highly sensitive digital imaging instruments (CCD
cameras) or X-ray film.
The light intensity produced directly correlates with the amount of antigen present on the membrane, enabling semi-quantitative analysis. Less sensitive but simpler systems include chromogenic substrates, which produce a visible colored precipitate.
02. Fluorescent Detection
Fluorescently tagged antibodies are detected using specialized imaging
instruments capable of capturing specific emission wavelengths. The key
advantage of fluorescent western blot detection is the ability to perform
multiplexing - detecting two or more proteins simultaneously on a single blot,
often including a housekeeping protein for normalization.
For this technique to be effective, specialized low-fluorescence PVDF membranes must be used to minimize high background interference.
Western Blot Analysis: Troubleshooting for Reliable Results
For beginners in Western blot, mastering troubleshooting is often the biggest hurdle. Many observed problems during detection originate from flaws in the initial sample handling or blocking steps. The following guide addresses common failures encountered during Western blot analysis.
| Problem Observed | Possible Causes & Context | Solutions and Protocol Adjustments |
|---|---|---|
| High Background / Dark Blot |
|
|
| Weak or Absent Signal |
|
|
| Multiple Bands or Smearing |
|
|
Table 02: Detailed Troubleshooting Guide for Western Blot
Analysis
NOTE: It is a common error to try to compensate for a high background by drastically reducing the secondary antibody concentration. However, if the root cause is that the initial lysis buffer lacked inhibitors, the protein is already degraded, causing smearing, or if the wrong blocking buffer was used, causing non-specific binding to Casein, no amount of antibody adjustment will yield a clean result. The most critical aspect of the Western blot protocol is the meticulous control of the sample and buffer chemistries at the very beginning.
Advanced Applications and Conclusion
The Western blot method remains a flexible and evolving technique, essential
across diverse fields from cancer research and immunology to drug development.
Advanced Applications
- Multiplexing and Normalization: The rise of digital imagers
and fluorescent western blot detection methods has facilitated multiplexing,
allowing researchers to simultaneously detect a target protein and an internal
loading control (e.g., GAPDH) on the same membrane. This significantly improves
the accuracy of semi-quantification in Western blot analysis. Total protein
normalization, often using fluorescent dyes to stain all transferred proteins,
is increasingly replacing single-housekeeping controls for more robust
quantitative results.
- Protein Modification Studies: The ability to specifically detect modifications like phosphorylation, acetylation, or ubiquitination makes the Western blot indispensable for understanding dynamic signaling pathways. Strict adherence to the rules regarding blocking agents (using BSA for phospho-antibodies) is paramount in these experiments.
The Western blotting technique is a powerful testament to combining physical
separation with immunological specificity. By rigorously following the
prescribed western blot steps, paying particular attention to sample integrity,
optimal loading, strategic membrane selection, and the chemical specifics of the
reagents, researchers can ensure that their western blot analysis delivers
reliable, impactful, and reproducible results, upholding its role as a
fundamental tool in the laboratory.
Faq's
01. Why is a fresh protease and phosphatase inhibitor cocktail essential for sample preparation?
They are critical to prevent the breakdown (proteases) or modification (phosphatases) of the target proteins after cell lysis. Their inclusion preserves the integrity and phosphorylation status of the proteins, preventing artifacts like smearing.
02. What is the key difference between PVDF and nitrocellulose membranes, and when should I choose one over the other?
PVDF is mechanically stronger and has a higher binding capacity, making it ideal for stripping and re-probing or for detecting low-abundance proteins. Nitrocellulose has lower autofluorescence, making it a better standard choice for fluorescent detection.
03. Why is the primary antibody dilution the most critical optimization factor for clean results?
The primary antibody determines the specific binding to your target. If the concentration is too high, it leads to excessive non-specific binding and a dark, high-background blot. Too low, and the signal will be weak or absent.
04. What is the function of the blocking step, and why is the choice of blocking agent important?
Blocking saturates the membrane's empty surface with inert protein (like milk or BSA) to prevent antibodies from binding non-specifically to the membrane itself, which significantly reduces background noise. BSA is often preferred over milk for detecting phosphorylated proteins to avoid interference from casein.
05. What is the purpose of the 'housekeeping protein' (e.g., GAPDH) or total protein normalization in Western blot analysis?
These are used as loading controls to ensure that an equal amount of total protein was loaded and successfully transferred into every lane, allowing for accurate semi-quantitative comparison of the target protein's relative abundance across different samples.